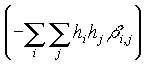
XTALDRAW
Online Reference Manual
This manual and the software described in it are copyrighted with all
rights reserved. Under the copyright laws, this manual or the software may not be copied
in whole or in part, without written consent of Bob Downs or Kurt Bartelmehs. The same
proprietary and copyright notices must be affixed to any permitted copies.
All Rights Reserved.
Command Reference
Atom Attribute Menu
Screen Center Menu
Interatomic Geometry
Video Mode Menu
Color and Shading Attribute
Sub-Menu
Data Input
Standard Setting
Non-standard Setting
Space Group Generator Input
Thank you for your interest in the program XTALDRAW . The word ''XTALDRAW'' is an abbreviation for ''Crystal Draw''. The version you are currently using is a registered shareware version of the software. As a registered user, we ask that you help us detect and determine any bugs that might exist in the software. In addition, if you think of ways that we can improve the software by making it easier to use or more useful to you, or even if you just have comments, we would appreciate hearing from you. You can contact either of us at the addresses located in this manual under Technical Support. We would be very happy to hear what you think about the software and reference manual.
As a registered user of XTALDRAW, you will be notified by email when future versions of the software become available.
Description and System Requirements
XTALDRAW is a 32 bit DOS based computer graphics program that displays and manipulates and analyzes ball and stick, polyhedral, and thermal ellipsoid colored drawings of any crystal structure or molecule. Even though it is not a Microsoft Windows
(RTM) application, it can be executed from within Windows using a full screen DOS shell. The program is written in FORTRAN 77 and compiled with Microsoft PowerStation 1.1a and is linked with the Fastgraph(trademark) Graphics Library 5.0. It requires a SVGA graphics card capable of displaying 256 colors and a Microsoft compatible mouse. Because the program operates in 32 bit protected mode, the file DOSXMSF.EXE (included with program) is also required.From the file openxtal.exe:
To install the XTALDRAW program and sample data files onto your hard disk, download the file "openxtal.exe" from the web site www.geo.utexas.edu/crystal into a temporary folder or directory. Run or "double click" on the file "openxtal.exe" to extract the necessary files to install the program. If successful, you should find a file named "setup.exe" in this temporary folder. Run or "double click" on the file "setup.exe" to install XTALDRAW into the desired location. An alternative method of obtaining "openxtal.exe" is by anonymous ftp from the directory /public/xtal at the ftp site crystal.geo.utexas.edu.
In order to execute the program from a DOS prompt, go to the appropriate subdirectory (the default subdirectory is C:\XTALDRAW) and type ''xtaldraw'' followed by an "enter". The program will automatically display drawings according to the highest of the resolutions listed below that is possible with your graphics card. Alternatively, the screen resolution can be specified by including a command line argument.
Command Line Argument |
Resolution (if possible) |
1 |
1024 x 768 pixels and 256 colors |
2 |
640 x 480 pixels and 256 colors |
3 |
800 x 600 pixels and 256 colors |
For example, to execute the program in 640 x 480 pixels and 256 colors, type "xtaldraw 2". This option is useful for displaying the program using an LCD projector. For more information about video modes, see Video Mode Menu (Alt v) described in the manual.
To run the program from within Microsoft Windows(RTM), open a full screen DOS shell and follow the DOS instructions or double left-click on the file ''xtaldraw.exe'' from within the File Manager or Windows Explorer.
Entering a data file to display
By default, the program title, version, and an introductory image will be displayed along with a box in the bottom of the screen that contains a message requesting the name of a data file. You may enter either the name of one of the data files provided with the software or one of your own data files. Data files automatically recognized by Xtaldraw have the file extension "xtl". If you have a Microsoft compatible Mouse properly connected to your computer, you can strike the F1 key to obtain a listing of all the files that have this extension within the current subdirectory. To make a selection, double left-click the mouse on the desired filename. For additional details see the Ctrl d option described in the Command Options section of this manual.
After selecting or entering the desired filename, the program will then calculate and display a ball and stick drawing of the structure. If the structure represents a crystal, then the c* axis is perpendicular to the screen, increasing toward the viewer, and the b axis is parallel to the horizontal direction of the screen, increasing from left to right with the center of the unit cell, [1/2 1/2 1/2], at the center of the screen. If the structure is a molecule, then the z axis is perpendicular to the screen and the y axis is horizontal, with the center of mass, [COM], placed at the center of the screen. For additional details about the default view, see the Ctrl Home option described in the Command Options section of this manual.
Once the drawing of the structure has been displayed, you may invoke any of the following options by striking the appropriate key or keystroke combinations represented in bold type. The mouse options require a Microsoft compatible mouse that is correctly connected and configured to the computer.
Alt a - to open the Atom Attribute Menu (see below).
Alt b - to turn on and off the lines or rods that represent the bonds.
Alt c - to open the Screen Center Menu (see below).
Alt d - to open a DOS shell from within the program. This command is useful for changing directories from within the program to access alternative data file directories. To exit the shell and return to the program, type ``exit'' followed by striking the enter key.
Alt e - to alter the probability (%) that is represented by the ellipsoids that are generated from advanced data sets. If no ellipsoids are currently drawn, this option will render ellipsoids for all atoms in which the appropriate data exists in the advanced data set. The default probability is 99.99999%. If the probability is entered as 0, then the isotropic temperature factors are drawn at a scale to make them approximately the same size as the ellipsoids at a probability of 99.99999%.
Alt f - to specify the face pole, (h,k,l) down which you want to view the structure. Three integers or real numbers can be entered. If you want to view the structure down a*, b*, or c* then you can type a, b or c. After striking this key, even if no numbers are entered, the viewing vector will be given with respect to reciprocal space coordinates (see Strike any other key). If the unit cell orientation figure is drawn (see Ctrl u), then the figure is drawn in reciprocal space.
Alt g - to switch between a ball and stick drawing and a polyhedral rendering of all cation coordinated polyhedra in the structure.
Alt i - to change the current rotation increment (unit of degrees). The default increment is 5o.
Alt l - to alter the display limits with respect to the a, b, and c axes. By default, the display limits include the regions (0 ® 1)a, (0 ® 1)b and (0 ® 1)c. All Cations within the display limits are drawn along with their coordinated anions. You may enter one, two, three, or six numbers at the prompt. The modification to the display limits is then determined according to the methods outlined in the following table. If the modification limits are set to zero, then the default limits are restored.
Option |
a |
b |
c |
default |
(0 ® 1) |
(0 ® 1) |
(0 ® 1) |
1 |
(0 - n) ® (1 + n) |
(0 - n) ® (1 + n) |
(0 - n) ® (1 + n) |
2 |
(0 - nl) ® (1 + nu) |
0 - nl) ® (1 + nu) |
(0 - nl) ® (1 + nu) |
3 |
(0 - na) ® (1 + na) |
(0 - nb) ® (1 + nb) |
(0 - nc) ® (1 + nc) |
6 |
(0 - nla) ® (1 + nua) |
(0 - nlb) ® (1 + nub) |
(0 - nlc) ® (1 + nuc) |
Alt n - to view program support information.
Alt o – to use the mouse to alter the position of the image with respect to the screen center.
Alt p - to alter the perspective or depth of the drawing. The default perspective is 0.004. A perspective value of 0.0 means no perspective.
Alt s - to turn on and off the shading mode. When the shading mode is turned on, a 3-dimensional shaded rendering will be constructed for the current image.
Alt t - to enter the number of times to translate or repeat the unit cell of a crystalline structure in each direction. If one number, (n), is entered, then the translation will be carried out n times along the ± a, ± b, ± c axes, respectively. If three numbers (na, nb, and nc) are entered, then the translation is carried out na times along ± a, nb times along ± b, and nc times along the ± c axis. It has no physical meaning for molecules.
Alt u - to turn on and off the unit cell outline.
Alt v - to open the Video Mode Menu (see below).
Alt w - To turn off and on the wire rendering mode for atoms drawn as spheres.
Alt x - to specify the lattice plane (h,k,l) cross section view of the structure. This gives a view of the structure with the direction of the face pole (h,k,l) oriented vertically and the user looking at the planes of atoms that give rise to X-ray diffraction. Using the or ¯ keys will scroll two parallel lines up and down the screen where the distance between these lines is equal to the d-spacing for the plane.
Alt z - to specify the zone [u,v,w] down which you want to view the structure. Three integers or real numbers can be entered. If you want to view the structure down a, b, or c then you can type a, b or c. If no numbers are entered, the viewing vector will be given with respect to direct space coordinates (see Strike any other key). If the unit cell orientation figure is drawn (see Ctrl u), then the figure is drawn in direct space.
up arrow- to rotate the image clockwise around an axis that runs from left to right through the center of the screen by the amount specified as the current rotation increment.
down arrow- to rotate the image counterclockwise around an axis that runs from left to right through the center of the screen by the amount specified as the current rotation increment.
left arrow- to rotate the image counterclockwise around an axis that runs from top to bottom through the center of the screen by the amount specified as the current rotation increment.
right arrow- to rotate the image clockwise around an axis that runs from top to bottom through the center of the screen by the amount specified as the current rotation increment.
Page Up - to rotate the image counterclockwise around an axis that runs perpendicular to the screen by the amount specified as the current rotation increment.
Page Down - to rotate the image clockwise around an axis that runs perpendicular to the screen by the amount specified as the current rotation increment.
plus or equal - to enlarge the size of the image.
dash or minus - to reduce the size of the image.
Ctrl a - Saves the current values of selected parameters in a file called XTALPARM.INI. The program then uses this file to obtain the default values of these parameters. The program default values can be re-established by deleting the file or resetting the parameters in the usual way and striking ctrl-a again. The parameters that are stored in this file include:
An additional file, ATOMATTR.INI, is also created that saves the current colors and radii of the atoms as last defined in the Atom Attribute Menu. In the case of duplicate atom names, the color and radius of the last one listed in this menu are set as the default values for that atom. Program defaults are restored by deleting this file.
Ctrl b - to switch between drawing and not drawing the borders of the polygons.
Ctrl d - to enter the name of a data file. By default, if the data file ends with the file extension ".xtl", then only the filename is required. When a wild character ''* '' is entered within the name, a listing of all files within the current subdirectory that satisfy the condition are displayed. Alternately, you can strike the F1 key to obtain a listing of all the files of the form ''*.xtl'' within the current subdirectory. If you have a Microsoft compatible Mouse properly connected to your computer, a selection can be made by double left-clicking the mouse on the desired filename. Furthermore, you can repeatedly strike the F2 key to display the names of up to five previously entered data files.
Ctrl g - to save the current image in GIF file format. By default, the file is named plotx.gif, where x is an integer between 001 and 999 and is automatically incremented. Optionally, any standard DOS filename can be entered. The default size of the file is equal to the current screen resolution, but can be modified by striking Ctrl i to set a scaling parameter. Satisfactory results are obtained when the GIF file is imported into commercially available programs such as PhotoShop. From there you can annotate the figure and/or send it to a printer, import into a web page or make a postscript file .
Ctrl i - to set the image file scale. The number entered is used to scale subsequent saved images in a pixel resolution that is equal to the current screen resolution times this number. Note that an image file scale equal to one (the default) at a resolution of 1024x768 pixels would produce an output image of little more than 3 inches wide on a 300 dpi laser printer. Increasing the image file scale results in larger, better quality output without bitmap scaling using a third-party application such as PhotoShop. Decreasing the image file scale is very useful for generating images to place in papers or web pages.
Ctrl n - to view the image generated from the next data set in the current data file.
Ctrl p - to save the current image in PCX file format. By default, the file is named plotx.pcx, where x is an integer between 001 and 999 and is automatically incremented. The default size of the file is equal to the current screen resolution, but can be modified by striking Ctrl i to set a scaling parameter. Satisfactory results are obtained when the PCX file is imported into commercially available programs such as Paint. From there you can annotate the figure and/or send it to a printer or make a postscript file.
Ctrl q or F3 - to quit the program.
Ctrl s - to control the size of the viewing dimensions. The size is defined as the distance between the left and right sides of the screen and is in the same units as the cell parameters of the currently displayed image. The value entered is used for all subsequent data sets during the current program session.
Ctrl u - to turn on and off the unit cell orientation figure. This is a small figure showing the unit cell vectors with their associated labels. The unit cell vectors can be displayed in direct or in reciprocal space (See alt z (direct space) or alt f (reciprocal space)). The vectors drawn on or out of the screen are drawn as solid lines whereas vectors drawn into the screen are represented as dashed lines. To change the position of this figure, first ``capture'' it by clicking the left mouse button on the origin of the figure. Then re-position the mouse to the desired location and click the left mouse button again.
Ctrl v - to enter the name of a PCX of GIF file. The extension ".PCX" or ".GIF" must be included in the file name. The image file is displayed until a key is struck thereby returning the original image. However, you can toggle back and forth between the image file and the original image by pressing the spacebar. The pressing of any other key takes you out of this particular option
Ctrl w – to turn on and off the mouse control window. When on, a small colored band appears around the edge of the screen. The currently displayed image can be rotated using the mouse by left-single-clicking in this area on the side representing the direction of rotation. The automatic rotation (i.e. animation) mode is activated if the mouse is left-double-clicked on the extreme outer edge of this band. If in the shaded mode (see alt-s), best results are obtained if perspective is set equal to zero (see alt-p). The direction of rotation can be changed dynamically by moving the mouse in and out of this outer band. The animation mode is deactivated by left-single-clicking in the area within the band. A second left-single-click will turn off the mouse control window.
Ctrl x – to enter a filename that will save the names and coordinates of all atoms enclosed within the unit cell in ASCII format
Home - returns the image to the default size and orientation.
Ctrl Home - to save the current image orientation setting as the default for the current session.
F10 - to save the current image coordinates to an ASCII file in Xtaldraw data file format.
Esc - to interrupt any option or to redraw the image.
Strike any other key - to display Title Line 1 of the current data set on the top line of the screen. The coordinates of the direction vector down which you are viewing the structure will be displayed as integers in the upper right hand corner if the individual coordinates are smaller than 999.
Right-click mouse - to turn on and off the visibility of the mouse icon.
Left-click mouse - anywhere on an atom to display the atom's name, id number and atomic coordinates along the bottom of the screen. If you left click on a polyhedral representation (alt-g), then the volume of the polyhedron will be displayed along with the name and coordinates of the central atom. By default the atomic coordinates are given with respect to the D = {a, b, c} basis or vector ''space''. By left-clicking on the letter ''D'', the atomic coordinates (and all subsequent coordinates) will be given with respect the C = {i, j, k} Cartesian basis. By left-clicking on the letter ''C'', the atomic coordinates will be re-displayed with respect to D.
Delete - to turn on and off the atom delete mode. When this mode is on, a left-click mouse anywhere on an atom will remove that atom from the set of displayed atoms. Striking any other key will automatically turn the atom delete mode off. To recover the deleted atoms strike alt-l and enter 0.
Click and drag mouse - to draw a box over the current image. Once the box is drawn, strike the Delete key to delete the contents of the box or strike the Insert key to crop or delete the contents outside of the box. To eliminate the box, strike any key.
This menu displays the current attributes of each unique atom in the unit cell. The attributes of an atom are defined by name, color, radius, and whether the atom is represented as a sphere, ellipsoid, polygon, or not drawn at all. For Cations, it also includes the bonded-radius. Other image attributes include the bond color and radius (applicable only in the 3-D shading mode), the border color that outlines the atoms, polygons and ellipsoids, the color and radius of the unit cell outline, and finally the background color. In order to change any of the image attributes, use the mouse or arrow keys to position the white box over the attribute to be changed, then follow the directions that appear at the bottom of the screen. All display changes will take effect upon exiting the menu.
Color - In order to change the current display color for an atom, left-click or right-click the mouse, or strike the + or - keys, to step forward and backward, respectively, through sixteen possible color choices. The color that is currently displayed will be the display color for that atom. Alternatively, strike Alt-c to open the Color and Shading Attribute Sub-Menu (see below) [Note that currently Alt-c works only when a Microsoft compatible mouse is detected and has a different effect outside of the Atom Attribute Menu].
Size - In order to change the radius of the circle that represents the spherical rendering of an atom, left-click or strike Enter. At the prompt, either enter a number that will represent the radius of the sphere or at any time strike Esc to quit this option. The default radius is computed as 0.4 times the Shannon and Prewitt (1969) radius.
Sphere - In order to display the atom as a sphere, left-click mouse, or strike Enter, until the word ''on'' appears in the column.
Ellipsoid - In order to display the thermal ellipsoid of an atom, left-click the mouse or strike Enter, until the word ''on'' appears in the column. If there is no data provided to construct an ellipsoid, then ''na'' will appear in the column. Upon exiting the menu, the thermal ellipsoid for the atom will be displayed for structures with advanced data sets. If the thermal ellipsoid cannot be displayed, for instance, if it has negative eigenvalues, then by default the atom will be drawn as a sphere.
Polygon - Cations only. In order to display cation coordinated polyhedra, left-click the mouse or strike Enter, until the word ''on'' appears in the column. If a polygon cannot be constructed then ''na'' will appear in the column. Upon exiting the menu, the polygon will be displayed in the color of the central cation. If the cation is not contained within the polygon, then by default the cation will be displayed as a sphere with the coordinating atoms not drawn.
Bond - Cations only. The number in this column represents the bonded-radius of the cation given in the data set for the structure. The bonded-radius defines the maximum distance to bonded atoms. In order to change the bonded-radius of the atom, left-click the mouse or strike Enter.
No Display - In order to NOT display the atom at all, left-click mouse button or strike Enter until the word ''on'' is removed from all the columns labeled sphere, ellipsoid and polygon.
Esc - To exit the menu.
This menu allows you to assign any position to be located at the center of the screen. In this menu all the unique atoms are enumerated in a list, along with the default positions [1/2 1/2 1/2] or [COM], special position [0 0 0], and the general position [X Y Z]. The current screen center coordinates are displayed at the bottom of the menu with the item in the list that corresponds to the current screen center coordinates highlighted in a different color.
To place a new position at the screen center, enter the number from the list that corresponds to the desired position. For instance, if you want to place an atom or a special position at the center of the screen, enter the number from the list that corresponds to that atom or special position. If you want to place an unlisted position at the center of the screen, enter the number corresponding to the general position [X Y Z]. At the prompt, enter the three coordinates that define the desired position.
To leave this menu, strike Esc. The desired position will be placed at the screen center for the duration of all other program command options.
Using Xtaldraw, it is possible to display interatomic distances and angles, polyhedral volumes, mean tetrahedral and octahedral quadratic elongation and angle variances (Robinson et al., 1971). The following actions require left clicking the mouse on the desired atoms.
Bond Lengths – to obtain the interatomic distance between any pair of atoms, left click on the first atom and then double left click on the second atom. A box appears in the lower left indicating the names of the two atoms and the distance between them.
Bond Angles – to obtain the interatomic angle between any three atoms, left click on the three atoms and then left click on the atom that subtends the angle. A box appears in the lower left indicating the names of the three atoms and the angle between them.
Cluster Calculation – to obtain the interatomic geometry of a central atom and its coordination sphere, double left click on the central atom or polyhedron. A screen will appear that enumerates the names, interatomic distances and angles of all coordinating atoms. If the central atom is a cation, the polyhedral volume will be displayed. If the polyhedron represents a tetrahedron or octahedron, then the angle variance and mean quadratic elongation will be displayed (Robinson et al., 1971). Strike any key to return to image display.
To clear or interrupt your atom selections, left click the mouse in a empty area of the screen.
This menu allows the selection of the following alternative video modes:
1024 ´ 768 pixel resolution ´ 256 colors
640 ´ 480 pixel resolution ´ 256 colors
600 x 800 pixel resolution x 256 colors
The capability of obtaining these modes is a function of your graphics card and monitor, and consequently certain modes may not be available on your computer. When the program first starts it chooses the video mode with the highest resolution as the default video mode.
Color and Shading Attribute Sub-Menu (Alt c)
This is a sub-menu invoked from the Atom Attribute Menu by striking alt-c when the cursor is over a color option for an atom. The menu displays the current red, green and blue color values for the selected color. The shine and gloss attributes used when the shading mode is turned on are also indicated (see Alt s in the Command Options section of this manual.) The values of these attributes can be changed by positioning the cursor at the appropriate part of the slide and left-clicking the mouse.
The direction of the light source is altered by positioning the cursor over the desired part of the shaded sphere and left-clicking the mouse. This direction affects the shading of spheres and polygons, and the position of the pole in the wire rendering of atoms.
To leave this menu, strike ESC.
In order for the program to compute and display a crystal or molecule, there must exist a data set within an ASCII file as outlined in Table 1. An example data set for the mineral pyrope is presented in Table 2. To construct an ASCII file, you can use the editor of your choice. Each individual line of a data set is unformatted, but must be entered in a particular order. The first three lines of a data set must contain three title lines that can be up to 80 characters in length. The first line, Title Line 1, can be displayed at the top of the image.
Table 1. General format of basic XTALDRAW data file.
* * * Top of File * * *
Title Line 1
Title Line 2
Title Line 3
a b c alpha beta gamma space-group-code
atom id
x y z coordination-sphere-radius
Anions
atom id
x y z
atom id
x y z
end
.
.
.
Additional data sets (optional)
.
.
.
Stop
* * * End of File * * *
The fourth line of a crystal data set must contain the six unit cell parameters (a, b, c,
a, b, g ) and a space-group-code. The units of measure represented by a, b, and c define the scale of the drawing. The space-group-code is given as either the standard space group symbol or the space group number, as defined in Table 6.2.1 of the International Tables for Crystallography, Volume 1 (Henry and Lonsdale, 1965). In the example presented in Table 2, the space-group-code is given as Ia3d. For a detailed discussion of space-group-code input, see the Space Group Input section of this manual. If the data set is for a molecule, then ``1 1 1 90 90 90 P1'' should be entered on this line.Table 2. Example of a basic data set for XTALDRAW
* * * Top of File * * *
pyrope: Novak and Gibbs, Am Min 56 (1971) 791-825
The crystal chemistry of the silicate garnets
natural sample, Py
11.459 11.459 11.459 90 90 90 Ia3d
Si
.375 0 .25 2
Al
0 0 0 2.5
Mg
.125 0 .25 3
anion
O
.03285 .05015 .65335
END
* * * End of File * * *
Finally, specific information about the type and position of the atoms contained in the structure or molecule must be entered in the data set. The program can recognize two categories of atoms, Cations and anions. In the strict chemical sense, a cation is an atom with a positive ionic charge while an anion is an atom with a negative ionic charge. By default, the program will consider all atoms input before the optional code word, ``anion'' or ``anions'', to be Cations, while those atoms input following the code word are considered to be anions (Tables 1 and 2). Regardless of ionic charge, the program will treat any atom input as a cation to be at the center of a coordinated polyhedron that is only bonded to atoms considered as anions. In turn, the program considers atoms classified as anions only to be bonded to Cations. If there are no atoms to be considered as anions, such as in a molecular crystal, then simply omit the code word.
Two lines of atomic information are necessary for each atom in the data set. The first line must contain an atom identifier. When this is the chemical symbol, the program can assign a default color to the atom. The second line must contain the three positional parameters, x, y and z. If the atom is input as a cation, then a distance (given in the same units as the cell parameters) defining the coordination-sphere-radius, must also be included. For example, in Table 2 the Si atom is located at the position [0.375 0.0 0.25] and all anions, in this case O atoms, will be considered to be bonded atoms if they are located within 2Å from the Si atom position. If you wish to enter multiple data sets within a single data file, then each data set must be separated by a line containing the word ''end'' (Table 1).
The program also permits you to enter the anisotropic displacement parameters (ADPs) for an atom in order to draw its thermal ellipsoid. The program recognizes three different algebraic forms of ADPs through the inclusion of the code word ''aniso_'' on the line preceding the input of the first atom id, as indicated in Table 3. The particular algebraic form of the ADPs is defined by the last letter given to ''aniso_'' and this letter is case sensitive. The following indicates three possible code words and the algebraic forms represented by each:
anisoB: exp
anisob: exp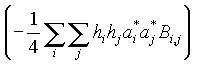
anisoU: exp .
.
Because the ADPs are the components of a 3´ 3 symmetric second rank tensor, you only need to enter the six unique tensor elements in the order (1,1), (2,2), (3,3), (1,2), (1,3), (2,3). The ADPs for the atom are entered on the second atomic information line after the positional coordinates as indicated in Table 3. An example of a data set for a cristobalite which includes input of the ADPs in the form ''anisoB'' is given in Table 4.
Table 3. The general format for an advanced XTALDRAW data set.
* * * Top of File * * *
Title Line 1
Title Line 2
Title Line 3
a b c alpha beta gamma space-group-code
aniso_
atom id
x y z _11 _22 _33 _12 _13 _23 coordination-sphere-radius
atom id
x y z _11 _22 _33 _12 _13 _23 coordination-sphere-radius
Anions
atom id
x y z _11 _22 _33 _12 _13 _23
atom id
x y z _11 _22 _33 _12 _13 _23
end
* * * End of File * * *
Table 4. An example of an advanced data set for XTALDRAW which includes input of the ADPs.
* * * Top of File * * *
cristobalite: Downs and Palmer, Am Min 79 (1994) 9-14
The pressure behavior of alpha cristobalite
This is room pressure data.
4.9717 4.9717 6.9223 90 90 90 P4_12_12
anisoB
Si
.30028 .30028 0 .0077 .0077 .00401 -.0003 -.0008 .0008 2
anion
O
.2392 .1044 .1787 .0244 .0086 .0062 -.0013 .0027 .0005
end
* * * End of File * * *
If ADPs are not available for input, the program will permit you to enter an alternate description of the thermal ellipsoid for an atom. This input requires the root-mean-square (rms) displacement of each principal axis and its angular relationship with the unit cell directions. In order for the program to recognize this form of input, you must include the code word ``anisoA'' or ``anisoa'' on the line preceding the input of the first atom id, as indicated in Table 5.
Table 5. The general format of the alternate, advanced XTALDRAW
* * * Top of File * * *
Title Line 1
Title Line 2
Title Line 3
a b c alpha beta gamma space-group-code
anisoa
atom id
x y z s s^a s^b s^c m m^a m^b m^c l l^a l^b l^c coordination-sphere-radius
Anions
atom id
x y z s s^a s^b s^c m m^a m^b m^c l l^a l^b l^c
atom id
x y z s s^a s^b s^c m m^a m^b m^c l l^a l^b l^c
end
* * * End of File * * *
Following the input of the atomic coordinates on the second atomic information line, the smallest principal rms displacement, s, is entered, along with the orientation angles, in units of degrees, of this principal axis with respect to the a, b and c direct basis vectors (s^a, s^b, s^c). Next, the intermediate length principal rms displacement, m, is entered, followed by its orientation angles (m^a, m^b, m^c), and then the largest rms displacement, l, and its orientation angles (l^a, l^b, l^c). Table 6 shows an example data set for calcite.
This option is also useful to display other types of oriented ellipsoids, such as the strain ellipsoid or optical indicatrix, within the context of the unit cell. Pleasing results can be obtained by placing the ellipsoid at [1/2 1/2 1/2]. An example data set is included in Table 6 for the strain ellipsoid of low albite between pressures of 0 and 3.78 GPa.
Table 6. Two examples of alternate, advanced XTALDRAW data sets showing input of the rms displacements of the thermal ellipsoid principal axes and their angular orientation for calcite and an example input data set to draw the strain ellipsoid for low albite.
* * * Top of File * * *
Calcite: Markgraf and Reeder, Am Min 70 (1985) 590-600
High-temperature structure refinements of calcite and magnesite
anisotropic refinement T=24C
4.988 4.988 17.061 90 90 120 R-32/c
anisoa
Ca
0 0 0 .1074 0 120 90 .1074 90 30 90 .112 90 90 0 2.5
C
0 0 .25 .107 0 120 90 .107 90 30 90 .1113 90 90 0 2
anions
O
.2567 0 .25 .0992 0 120 90 .1249 90 53.3 43.7 .179 90 51.2 133.7
end
strain ellipsoid P=0-3.78 GPa: Downs et al., Am Min 79 (1994) 1042-1052
The high-pressure crystal chemistry of low albite and the origin of
the pressure dependency of Al-Si ordering
8.1372 12.7867 7.1574 94.245 116.605 87.809 P1
anisoA
strain
.5 .5 .5 .2866 103 20.5 75 .3381 112.7 108.9 15 .9114 26.3 82.3 91.2 1
end
* * * End of File * * *
Sometimes ADPs have not been determined for all the atoms in a structure. For this reason, another form of data set exists that contains both atoms with ADPs and atoms without ADPs. The general form for this type of data set is given in Table 7. For a mixed data set, all atomic information for both Cations and anions, must be categorized according to the code words ''aniso_'' and ''iso''. Following the appropriate ''aniso_'' code word, the anions with ADPs are listed with the format described in the Advanced Data Set section. If there are Cations without ADPs, then the code word ''iso'' is entered next, followed by the basic atomic information for each of these atoms as described in the Basic Data Set section. Similarly, following the code word ''anions'', any anions with ADPs are listed following the appropriate ''aniso_'' code word, while basic atomic information on any anion without ADPs is listed following the code word ''iso''. An example of a mixed data set is provided in Table 8.
Table 7. The general format for a mixed XTALDRAW data set.
* * * Top of File * * *
Title Line 1
Title Line 2
Title Line 3
a b c alpha beta gamma space-group-code
aniso_
atom id
x y z _11 _22 _33 _12 _13 _23 coordination-sphere-radius
iso
atom id
x y z coordination-sphere-radius
Anion
aniso_
atom id
x y z _11 _22 _33 _12 _13 _23
iso
atom id
x y z
end
* * * End of File * * *
Table 8. An example of a mixed XTALDRAW data set.
* * * Top of File * * *
Poldervaartite: Dai, Harlow and McGhie: Am Min 78 (1993) 1082-1087
Poldervaartite, Ca(Ca.5Mn.5)(SiO3OH)(OH), a new acid nesosilicate from the
Kalahari manganese field, South Africa: Xtal structure and description.
9.398 9.139 10.535 90 90 90 Pbca
ANISOU
Ca
.15406 .49326 .39021 .009 .0101 .0084 -.0004 .0007 .0003 3
M2(Ca,Mn)
.51266 .3339 .43048 .0101 .0104 .0098 -.0022 -.0003 -.0004 3
Si
.32973 .21515 .65939 .0076 .0075 .0069 .0002 -.0002 -.0004 2
iso
H1
.257 .074 .117 1
H2
-.084 .588 .173 1
anions
ANISOU
O1
.2518 .3502 .5735 .028 .0104 .014 .0046 -.0084 -.0005
O2
-.0559 .359 .4368 .0092 .0145 .0117 -.0019 -.0002 -.0042
O3
.102 .7101 .2829 .016 .0134 .0087 .0022 .0045 .0043
O4
.3965 .5492 .398 .0118 .019 .016 -.0012 -.0002 .0063
O5
.2019 .3986 .1893 .0146 .0145 .0129 .0044 .0018 -.0016
end
* * * End of File * * *
There is a line in the data file that must contain the six unit cell parameters and a code specifying the symmetry of the structure:
a b c
a b g space-group-code,where the space-group-code can be entered as either any one of the standard short space
group symbols or space group numbers as given in Table 6.2.1 of the International Tables
for X-ray Crystallography, Volume 1 (Henry and Lonsdale, 1965). The lattice-type element
of the space group symbol must be capitalized while the other symbol components must be in
lower case. For example, the space-group-code for space group # 230, i.e. Ia3d, can
be entered either as Ia3d or 230. Screw axes are designated with an underscore. For
example, the space-group-code for space group # 144, i.e. P31, can be entered
as either P3_1 or 144. Bar operations are designated with a minus sign. For example, the
space-group-code for space group # 147, i.e. P![]() , can be entered as
either P-3 or 147. For cubic and hexagonal crystals, the program recognizes either the
standard full or standard short space group symbols. For example, the space-group-code for
space group #221, i.e. P4/m
, can be entered as
either P-3 or 147. For cubic and hexagonal crystals, the program recognizes either the
standard full or standard short space group symbols. For example, the space-group-code for
space group #221, i.e. P4/m![]() 2/m, can be entered as either P4/m-32/m,
Pm-3m or 221. The program automatically distinguishes between first and second settings
for the monoclinic crystal system, and between rhombohedral and hexagonal settings for the
rhombohedral classes, by examining the values input for a , b and g
2/m, can be entered as either P4/m-32/m,
Pm-3m or 221. The program automatically distinguishes between first and second settings
for the monoclinic crystal system, and between rhombohedral and hexagonal settings for the
rhombohedral classes, by examining the values input for a , b and g
A space group setting that is not listed as a standard setting in the International
Tables for X-ray Crystallography (Henry and Lonsdale, 1965) can be obtained from one that
is standard through a transformation. The program recognizes the symbols for various
non-standard settings in the orthorhombic crystal system, such as cab, bca, a![]() b, ba
b, ba![]() and
and ![]() ba,
provided that they are unique, and automatically computes the transformation. For example,
with space group # 62 the standard setting is Pnma. However, the non-standard settings
Pbnm, Pmcn, Pnam, Pmnb and Pcmn can all be input as the space-group-code representing
their respective non-standard settings.
ba,
provided that they are unique, and automatically computes the transformation. For example,
with space group # 62 the standard setting is Pnma. However, the non-standard settings
Pbnm, Pmcn, Pnam, Pmnb and Pcmn can all be input as the space-group-code representing
their respective non-standard settings.
When the symbol cannot be specified, you can input either the appropriate rotation matrix and/or a translation vector needed to compute the transformation. A transformation to an non-standard setting is indicated in the data file by preceding the standard setting space group symbol with an asterisk, * , or by changing the space group number to its negative value. The next lines in the data file must then contain the translation vector and/or a 3´ 3 rotation matrix. For example, the non-standard setting chosen for quartz by Kihara (1990) involves a translation of the origin of the standard setting by 2/3 along the z-axis. Input of this space group information could be given as
4.9137 4.9137 5.4047 90 90 120 *P3_221
0 0 .6666666666667
where 2/3 = .6666666666667 was input to ensure precision in the calculations. As another example, consider the P21/a non-standard second setting chosen for parawollastonite by Hesse (1984). This space group setting involves a rotation of the basis vectors (a'b'c') defined in the standard setting for P21/c (space group # 14) to the setting of c'b'a' (see Table 6.2.1 in the International Tables for X-ray Crystallography). Here the space group information could be entered as
15.409 7.322 7.063 90 95.3 90 *P2_1/c
0 0 1
0 1 0
1 0 0
where the rotation matrix is obtained from the change in the space group symbols. The difference in the order of the basis vectors indicates that the rotation maps a' to c', b' to b' and c' to a', thus defining the columns of the rotation matrix. If both a translation and rotation are required, the translation vector must be entered first.
When a space group with a non-standard setting cannot be easily constructed by a
transformation of the standard setting, the program permits you to input the matrix
representations of the space group generators, as discussed in Boisen and Gibbs (1990). In
order to input these matrices, enter a value of zero for the space-group-code. The next
line must contain the number of generators to be input. The next three or more lines of
the input file must contain the row elements of each 3´ 4
generator matrix. Following the matrix input, the next lines must contain the lattice
vectors with the 0 0 0 lattice vector given on the last line of the space group
information. For example, low albite is described with C![]() symmetry by Winter et al. (1977), so its space group information is entered
as
symmetry by Winter et al. (1977), so its space group information is entered
as
8.205 12.817 7.169 93.97 116.39 87.69 0
1
-1 0 0 0
0 -1 0 0
0 0 -1 0
.5 .5 0
0 0 0
where a single 3´ 4 generator is given, along with the lattice conditions for a C-centered lattice. As another example, consider the I2/c non-standard second setting chosen by Griffen and Ribbe (1976) to describe the mineral celsian. This space group information can be input as
8.622 13.078 14.411 90 115.09 90 0
2
-1 0 0 0
0 -1 0 0
0 0 -1 0
-1 0 0 0
0 1 0 0
0 0 -1 .5
.5 .5 .5
0 0 0
where two generators are input along with the lattice vector for an I-type lattice. Other examples can be found by examining the sample data files provided with the software.
For problems or suggestions contact either:
Dr. Kurt L. Bartelmehs
Department of Geological Sciences
The University of Texas at Austin
Austin, Texas 78712 USA
Voice: (512) 475-6754
Fax: (512) 471-9425
Email: [email protected]
Dr. Bob Downs
Department of Geosciences
Gould-Simpson Building
The University of Arizona
Tucson, Arizona 85721 USA
Voice: (520) 626-8092
Fax: (520) 621-2672
Email: [email protected]
We want to thank G.V. Gibbs and M.B. Boisen, Jr. at Virginia Tech for their support through NSF grants EAR-8803933 and EAR-9303589. Virginia Tech is also thanked for supporting this work with a Virginia Tech Outreach Grant for the Promotion of Science Among the Young People of Virginia, along with Media Services at Virginia Tech for donating computer equipment. We also thank R.M. Hazen and L.W. Finger at the Carnegie Institution of Washington for their support through NSF grant EAR-9218845. We wish to dedicate this program to Professors Jerry Gibbs and Monte Boisen, Jr. for their outstanding tutelage in the field of mathematical crystallography.
Internet users can obtain the unregistered shareware program package by anonymous ftp from the directory /public/xtal at the ftp site crystal.geo.utexas.edu or visit the web site at www.geo.utexas.edu/crystal.
Boisen, M.B., Jr. and Gibbs, G.V. (1990) Mathematical Crystallography, Volume 15, Reviews in Mineralogy, 406 pp. Mineralogical Society of America Washington D.C.
Dai, Y., Harlow, G.E. and McGhie, A.R. (1993) Poldervaartite, Ca(Ca.5Mn.5) (SiO3OH)(OH), a new acid nesosilicate from the Kalahari manganese field, South Africa: Crystal structure and description. American Mineralogist, 78, 1082-1087
Downs, R.T., Hazen, R.M. and Finger, L.W. (1994) The high-pressure crystal chemistry of low albite and the origin of the pressure dependency of Al-Si ordering. American Mineralogist, 79,1042-1052.
Downs, R.T. and Palmer, D.C. (1993) The pressure behavior of a cristobalite. American Mineralogist, 79, 9-14.
Griffen, D.T. and Ribbe, P.H. (1976) Refinement of the crystal structure of celsian. American Mineralogist, 61, 414-418.
Henry, N.F.M. and Lonsdale, K. (1965) International Tables for X-ray Crystallography, Volume 1, 558 pp. The Kynoch Press, Birmingham, England.
Hesse, K.F. (1984) Refinement of the crystal structure of wollastonite-2M (parawollastonite). Zeitschrift für Kristallographie, 168, 93-98.
Kihara, K. (1990) An X-ray study of the temperature dependence of the quartz structure. European Journal of Mineralogy, 2, 63-77.
Markgraf, S.A. and Reeder, R.J. (1985) High-temperature structure refinements of calcite and magnesite. American Mineralogist, 70, 590-600
Novak, G.A. and Gibbs, G.V. (1971) The crystal chemistry of the silicate garnets. American Mineralogist, 56, 791-825
Robinson, K, Gibbs, G.V. and Ribbe, P.H. (1971) Quadratic Elongation: Quantitative Measure of Distortion of Coordination Polyhedra. Science, 172, 567-570.
Shannon, R.D. and Prewitt, C.T. (1969) Effective ionic radii in oxides and fluorides. Acta Crystallographica, B25, 925-946
Winter, J.K., Ghose, S. and Okamura, F.P. (1977) A high-temperature study of the thermal expansion and the anisotropy of the sodium atom in low albite. American Mineralogist, 62, 921-931.
Pages modified on 05/04/01
Comments to [email protected]