Inorganic Chemistry, NUI, Galway
This worked example is EX1 and there are ten other practicals 3yr1 - 3yr10.
For each case TXT and HKL and GIF files are provided.
A worked example EX1
Start Oscail.
Use Change Directory ![]() to point Oscail at the directory containing EX1.TXT and EX1.HKL
to point Oscail at the directory containing EX1.TXT and EX1.HKL
The status line should show Jobname ex1 and the Current Directory you are using..
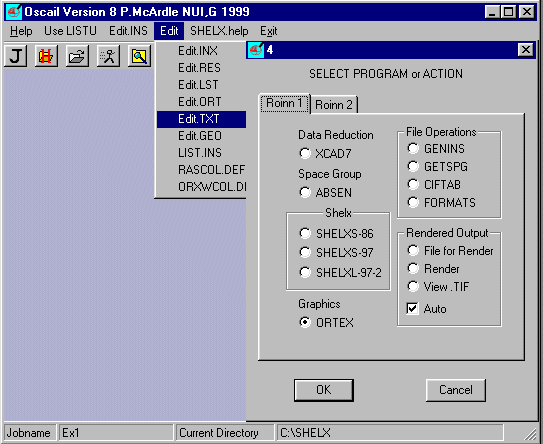
Select Edit and Edit.TXT
The file EX1.TXT contains the following information.
TITL EX1
Molecular formula C27 H20 O4 Fe2 Sn1
UNIT CELL 14.0650 11.7470 15.2140 90.000 105.930 90.000
CELL ESDS 0.00100 0.00200 0.00300 0.00000 0.01000 0.00000
CELL VOLUME 2417.2(3) �3
Write this information in your lab notebook.
On the basis that all non-H atoms occupy about 20 �3 the
molecular
formula in this case containing 34 non-H atoms would occupy about
670 �3. This divides into 2417 (CELL VOLUME) 3.6 times. Thus Z
the number of molecules in the UNIT CELL will be about 4. The crystal
system is Monoclinic with a beta angle of 105.93 deg.
Space Group possibilities can be examined using the program ABSEN.
Determination of the correct Space Group is one of the most
important steps in determining a crystal structure.
Select Run Job ![]() and ABSEN
and ABSEN
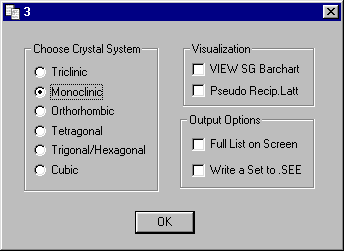
Select Monoclinic as the Crystal System (default)
The following table is produced.
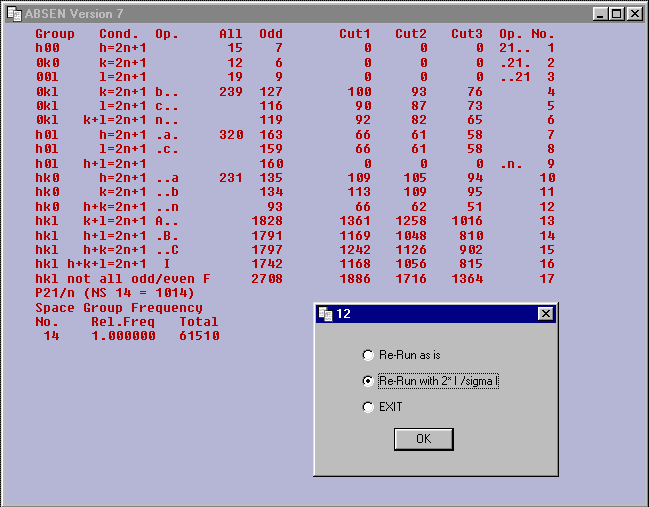
Examine the table.
The Space Group is clearly P21/n a non-standard setting of P21/c No.
14.
It can be selected using the number 1014.
The Space Group frequency information indicates that this is the
most common Space Group that was on the Cambridge Data Base at
the end of 2000.
Put P21/n and 1014 into your note book.
Choosing a space group is not always a simple matter but in this case
it is uniquely determined by systematic absences. The most important absence
is h0l h+l=2n+1. To generate a picture of the h0l layer in
reciprocal space do the
following. Select Re-Run as is and OK and then select Pseudo
Recip.Latt under
Visualization and OK click OK on Dialog 14 and Exit Absen.
Change the Jobname
to PH0L (that is PHzeroL) and run ORTEX. Using Atom Mode you get the following
picture
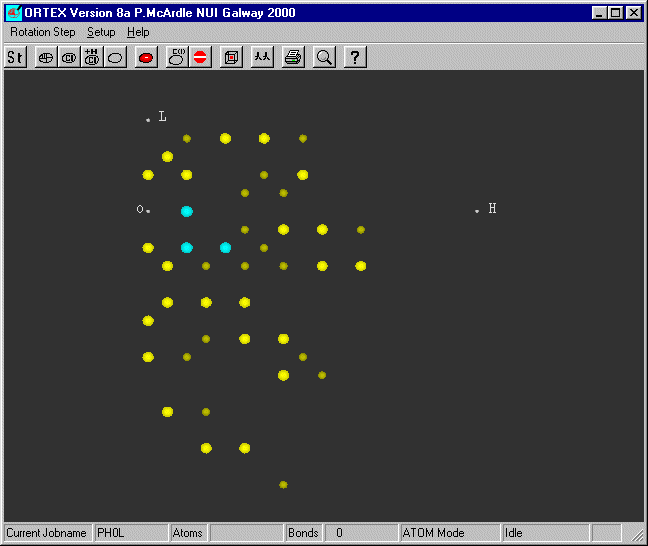
The brightest bigest spots are the most intense. Notice that spots on adjacent
levels
are never under each other as with L=1 all H are odd and with L=2 all H are
even.
Exit ORTEX and reset the Jobname back to EX1.
STEP 3
You are now ready to generate the input Files for SHELXS.
Select Run Job ![]() and GENINS
and GENINS
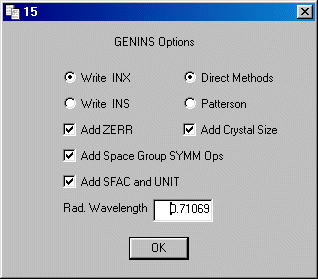
Select Write .INX and also Add Zerr,
Add Space Group SYMM Ops and Add SFAC and UNIT
Add a comment line to the files, this may be useful in the future.
Insert the UNIT CELL dimensions a b c alpha beta gamma from
your notes (EX1.TXT). Use the down cursor to move down.
UNIT CELL 14.0650 11.7470 15.2140 90.000 105.930 90.000
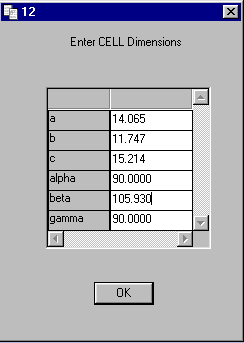
On Dialog 13 Enter ZERR
Set Z to 4 and the ESDS
to the values in EX1.TXT
CELL ESDS 0.00100 0.00200 0.00300 0.00000 0.01000 0.00000
From the Add Symm Ops default
Select Number on Dialog 11
Insert 1014 (the number of P21/n) on Dialog 10
From Add SFAC and UNIT
Insert the Molecular formula as given in EX1.TXT (the 1 after Sn is required)
C27 H20 O4 Fe2 Sn1
More information is now written to the screen including that
the mean atomic volume per non-H atom which at 17.8 is not
too bad and close enough to the 20 that was suggested in STEP 1.
When you return to Oscail
Select Run Job ![]() and SHELXS-97
and SHELXS-97
SHELXS-97 will now use Direct Methods to try to solve the structure.
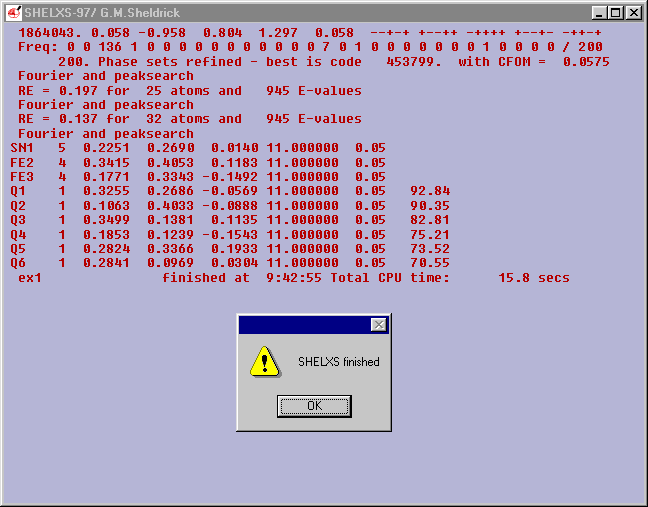
The best Combined Figure Of Merit (CFOM) is
0.0575.
It is a guide to the quality of the solution found.
0.0575 is satisfactory. It is difficult to generalize but the
following may be useful
a CFOM of >0.18 is unlikely to be correct
a CFOM of <.08 is o.k.
a CFOM 0f <0.01 could indicate trouble.
But no problems here the peak search found 3 heavy atoms and labelled
them as one Sn and two Fe with about 30 other atoms.
Click inside the SHELXS window to close it.
You can examine the SHELXS output in detail when you
return to Oscail using Edit.LST
STEP 4
What you do at this stage depends on your experience it would be normal
to delete rubbish and to start to refine 'good' atoms ![]() , but new-users
, but new-users
do the following:
Select Run Job ![]() and ORTEX (default)
and ORTEX (default)
If asked Use Existing .ORT ?
answer NO
to Use Defaults ?
answer YES
If Dialog 30 appears with .INS or .RES ?
select .RES
What now appears on the screen may look like a mess but
select rotate and spin the structure around and you will see
a five membered ring.
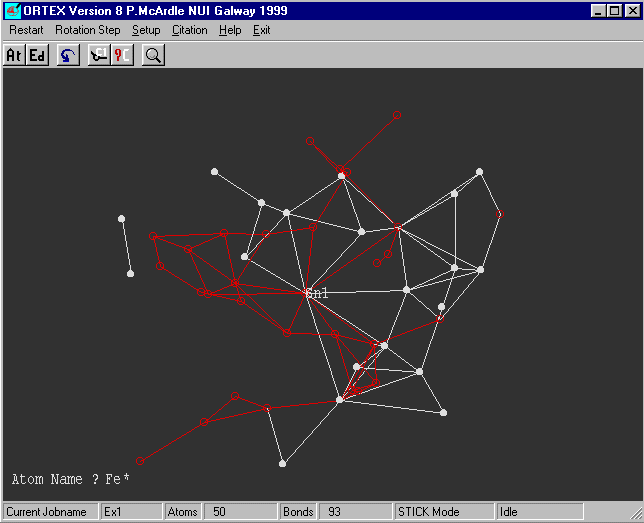
We know that the CFOM was o.k. and the Sn and Fe(s) look reasonable.
This together with some more 'good atoms' is enough for SHELXL-97/2.
Exit ORTEX
Having obtained a solution in STEP 4 the idea behind refining at this
stage is that
real atoms will refine to give reasonable thermal parameters and rubbish will give
poor thermal parameters. All atoms other than the Sn and Fe atoms are given the
atom type number of carbon (1).
Select Run Job ![]() and SHELXL-97/2.
and SHELXL-97/2.
Notice that the wR2 is dropping through the cycles.
answer Yes to Overwrite .INS ?
Select Edit.INS
The thermal parameter is in the last column and the 'atoms' with
parameters greater
than 0.10 are probably just rubbish. Delete the lines in Red and put ANIS
3 before
Sn1. ANIS 3 will allow the three metal atoms to refine anisotropically (as ellipsoids).
This will remove 'ghost peaks' which tend to appear around heavy atoms when refined
isotropically (as spheres).
The MOLE instructions are not required.
WGHT 0.100000
FVAR 0.13637
MOLE 1
ANIS 3
SN1 5 0.226249 0.267577 00.016342 11.00000 0.03291
FE2 4 0.346042 0.406012 00.121607 11.00000 0.03795
FE3 4 0.178050 0.335470 -0.150092 11.00000 0.04625
Q1 1 0.324649 0.2998930 -0.033997 11.00000 0.39306
Q2 1 0.024962 0.4256730 -0.157574 11.00000 0.17959
Q3 1 0.358723 0.0867860 00.094139 11.00000 0.29454
Q4 1 0.161066 0.1129700 -0.205832 11.00000 0.32711
Q5 1 0.326852 0.2983050 00.232021 11.00000 0.43936
Q6 1 0.285237 0.0971800 00.034347 11.00000 0.03731
Q7 1 0.139994 0.1960100 -0.174997 11.00000 0.06230
Q8 1 0.504608 0.3021610 00.063233 11.00000 0.04464
Q9 1 0.326816 0.0329210 00.113064 11.00000 0.43280
Q10 1 0.296678 0.304419 -0.153253 11.00000 0.05835
Q11 1 0.189732 0.510961 -0.161369 11.00000 0.05828
Q12 1 0.089445 0.240542 00.057026 11.00000 0.03657
Q13 1 0.244192 0.536331 00.060608 11.00000 0.03715
Q14 1 0.161799 0.116853 00.016950 11.00000 0.37455
Q15 1 0.259947 0.535667 00.159385 11.00000 0.04944
Q16 1 0.351887 0.250176 00.271245 11.00000 0.04657
Q17 1 0.375769 0.288576 -0.160103 11.00000 0.05671
Q18 1 0.141003 0.531732 -0.009313 11.00000 0.04511
Q19 1 0.131593 0.488191 -0.098591 11.00000 0.05230
Q20 1 0.439979 0.337379 00.087887 11.00000 0.04919
Q21 1 0.110924 0.102530 -0.192982 11.00000 0.06496
Q22 1 0.198965 0.390682 00.104992 11.00000 0.41035
Q23 1 0.265062 0.026211 00.098966 11.00000 0.05887
Q24 1 0.346672 0.309081 00.206695 11.00000 0.05925
Q25 1 0.208016 0.311947 00.134244 11.00000 0.49679
Q26 1 0.340277 0.508694 -0.019843 11.00000 0.32747
Q27 1 -0.002052 0.272061 0.172787 11.00000 0.04887
Q28 1 0.084082 0.292830 00.145002 11.00000 0.04032
Q29 1 0.368794 -0.121841 0.072809 11.00000 0.07232
Q30 1 0.245165 0.438261 -0.015010 11.00000 0.24886
Q31 1 0.293268 0.180800 -0.070845 11.00000 0.36132
Q32 1 0.409282 0.566419 00.129207 11.00000 0.06700
Q33 1 0.281415 0.359296 -0.050363 11.00000 0.30349
Q34 1 0.348582 0.055364 -0.012710 11.00000 0.07378
Q35 1 0.087470 -0.00050 -0.212377 11.00000 0.40814
Q36 1 0.336477 0.557178 00.045282 11.00000 0.05168
Q37 1 0.263745 0.392233 -0.134591 11.00000 0.34451
Q38 1 0.375668 0.374770 00.239078 11.00000 0.56961
Q39 1 0.309406 -0.083685 0.119520 11.00000 0.06808
Q40 1 0.463020 0.252230 00.233987 11.00000 0.46474
Q41 1 0.364424 0.552989 00.204016 11.00000 0.05813
Q42 1 0.116633 0.245358 -0.245414 11.00000 0.41070
Q43 1 0.366037 0.183805 00.132432 11.00000 0.57140
Q44 1 -0.059113 0.142311 0.046392 11.00000 0.07612
Q45 1 0.144898 0.399769 -0.090477 11.00000 0.41199
Q46 1 -0.068108 0.194979 0.130077 11.00000 0.06179
Q47 1 0.393722 -0.055587 0.003426 11.00000 0.07620
MOLE 2
HKLF 4
Save the file and exit PFE.
Select Run Job ![]() and SHELXL-97/2
and SHELXL-97/2
The wR2 value will now drop further.
answer Yes to Overwrite .INS ?
Select Run Job ![]() and ORTEX (default)
and ORTEX (default)
answer No to Use existing ex1.ORT ?
answer No to Use Defaults ?
answer YES to Use Covalent Radii for Bonds ?
On Dialog 2 Check Add Diff Map Qs and OK.
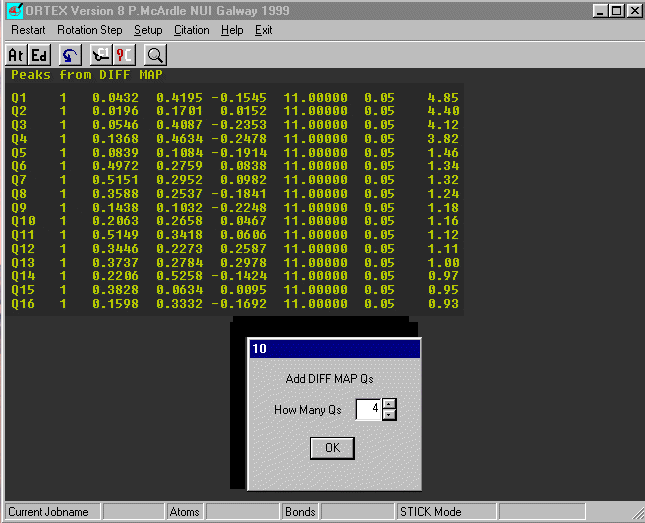
Select Use .INS (default) on Dialog 30.
The first 16 peaks in the Difference Fourier are now listed.
The last column is the peak height and notice there is a clear drop after
peak 4. Insert the number of large peaks (4) into Dialog 10 and OK.
A 'stick' picture of the molecule now appears on the screen.
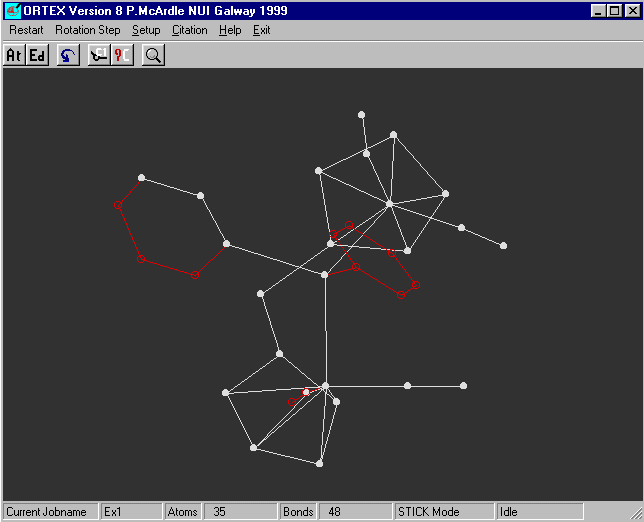
The arrow keys are used to rotate the molecule
for y Rotation use the left/right cursor keys, the up/down keys for z
and shift + left/right keys for x
If you rotate the molecule you will see that all looks reasonable.
It is now necessary to name all the atoms with reasonable names.
Number the Carbons from C1 up to C27 and the four Oxygens from O1 - O4.
The oxygens are on the CO groups attached to the Fe atoms.
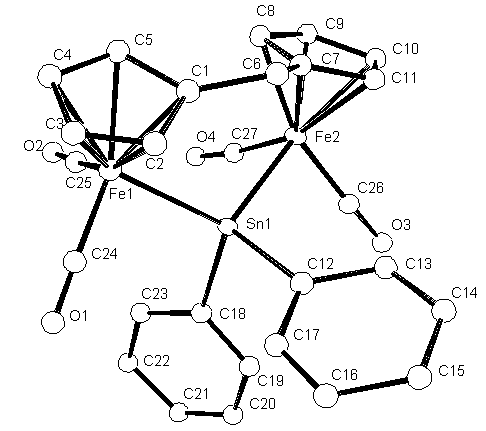
To rename the atoms select Edit ![]() on the
ORTEX (stick) Menu.
on the
ORTEX (stick) Menu.
You need to be careful. In Edit atoms are selected with the mouse
and the atom's name is shown on the bottom left. When an atom is
selected three actions are possible.
1. Click a blank area of the screen to clear the selector.
2. Press ![]() (or C on the keyboard) and you can change an
atoms name.
(or C on the keyboard) and you can change an
atoms name.
3. Press ![]() (or D on keyboard) to delete an
atom.
(or D on keyboard) to delete an
atom.
Change (rename) all of the Carbon and Oxygen atoms and then
Select Update (this will
overwrite the old
.INS file)
Select the Default options.
The result of the ORTEX operations are now written to EX1.INS
The Carbon atoms are last and sorted in numerical order.
Before this file is sent to SHELXL-97/2 use Edit.INS to
check that all atoms have Unique Names. Duplicate names will
cause SHELXL to fail. If you have made a mistake and have 2 C13s
say then just change one of them to C53 for the moment.
Select Run Job ![]() and SHELXL-97/2.
and SHELXL-97/2.
The wR2 factor will now decrease again. If the maximum and minimum
peaks in the Difference map are not greater than 1.0 (absolute value) then
you have found and refined all of the non-H atoms.
To add hydrogen atoms the SHELX HFIX command can be used.
Use Edit.INS and put in the following three lines after
FMAP 2
HFIX 43 -1.3 C2 C3 C4 C5 C8 C9 C10 C11
HFIX 23 -1.3 C6
ANIS
(where 43 is for sp2 type CH and 23 is for secondary CH2 the -1.3
asks for the H to have a thermal paramater 30% than its C and ANIS
will allow all non-H to refine anisotropically)
Select Run Job ![]() and SHELXL-97/2
and SHELXL-97/2
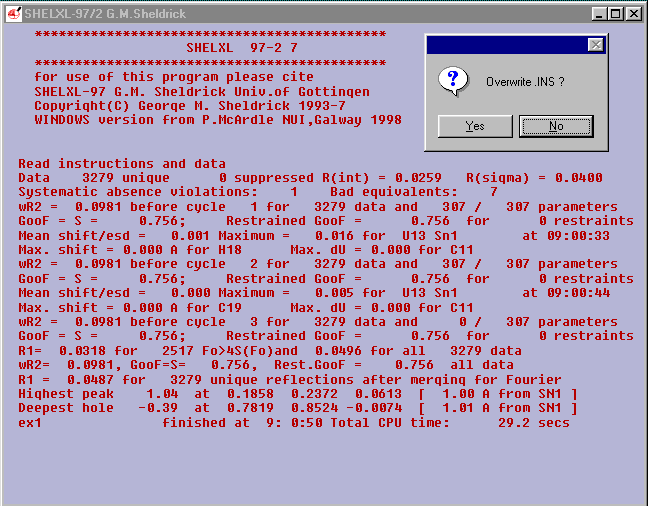
answer Yes to Overwrite .INS ?
EX1 should now be fully refined..
STEP 6
Select Run Job ![]() and ORTEX
and ORTEX
answer No to Use existing ex1.ORT ?
answer Yes to Use Defaults ?
Select Use .INS (default) on Dialog 30.
Select Rotate and get a good view Select Atom ![]() then Ellipsoids
then Ellipsoids ![]()
POS.L ![]() can be used to position atom labels.
can be used to position atom labels.
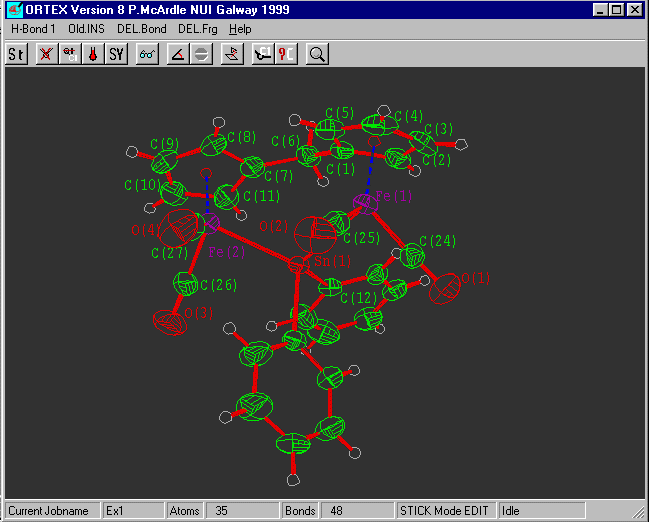
Select Hardcopy ![]() and print directly on
your printer or write HPGL or WMF for
and print directly on
your printer or write HPGL or WMF for
insertion into WORD etc.
Exit ORTEX
Inter Molecular Contacts
There are no expected inter molecular H-bonds in this case. It is possible to check for
C-H...O
interactions (which are currently popular) as follows. Since the contacts would only be
from oxygen
it is only necessary to search around these atoms.
Select Run Job ![]() and ORTEX
and ORTEX
answer Yes to Use existing ex1.ORT ?
Select Atom ![]() and Pac/Con
and Pac/Con ![]()
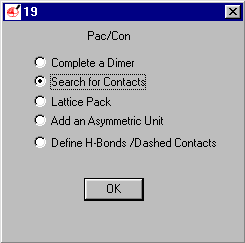
Select Search for Contacts (default)
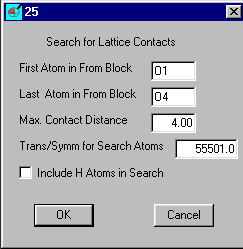
Insert O1 and O4 (case does not matter) as the atom range and set the max Contact distance
to 3.4 and OK
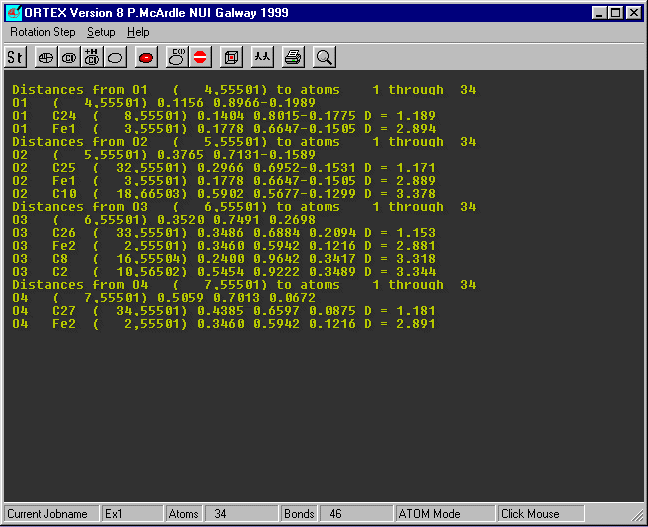
The contact distances are shown on the right the O...C distances less than
3.4 are shown and three of them
would merit further investigation.
Exit ORTEX
STEP 8
Building the Crystal Lattice
Select Run Job ![]() and ORTEX
and ORTEX
answer No to Use existing ex1.ORT ?
answer No to Use defaults ?
Check Add Unit Cell on Dialog 2 and
default on other Dialogs
Select Atom ![]() and Pac/Con
and Pac/Con ![]()
Select Lattice Pack and OK
The defaults will fill one cell.
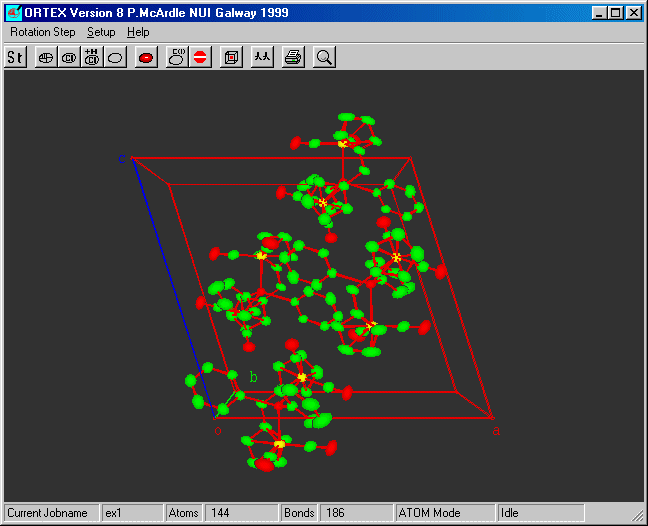
STEP 9
Tables of Data for Your Report
Add the command ACTA to the .INS file
after FMAP 2
and run SHELXL-97/2 again. This will generate .CIF and .FCF files.
From Oscail select Run
Job ![]() and CIFTAB
and CIFTAB
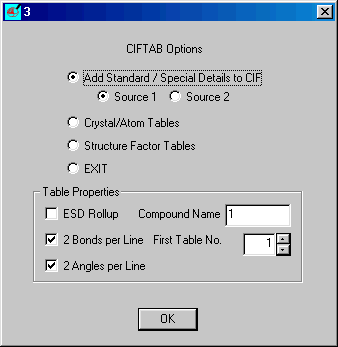
Select Add Standard etc. (default) and
insert the Compound Name (code) and click OK
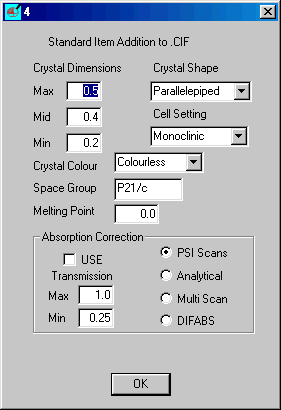
Insert the correct items and click OK
When dialog 3 returns select Crystal/Atom Tables
and OK
A .TEX file is now written. Start WORD and use Insert File
to insert the .TEX file. The addition of Greek characters superscripts etc.
can be done using a macro which may not work if you are on a LAN server. The
instructions for a standard PC setup are (taken from Oscail HELP).
The
DOT files supplied with Oscail are Galway2k.DOT or (Galway.DOT for WORD 6) and
they will be in \ortex. Find where WORD templates, DOT files, are stored on your
PC. In most cases this will be \windows\application data\Microsoft\templates
(you should see normal.dot there). Copy the DOT files from \ortex to this
directory. You can either use File/New in WORD and open a new document using
Galway2k.dot as a template or you can use, from an existing document, Tools
(expand down the dialog and underneath Macro
you will find Templates and Add-Ins
select this option and on the next dialog click add and select the Galway2k.DOT
or (Galway.DOT for WORD 6) check its box if necessary and OK.
Select Tools/Macro and Run the Galway macro
Greek characters etc. should appear..
Select Table 1 and use
Convert Text to Table (select comma) to format the (comma delimited)
table.
Repeat for the other tables.

Select a small number of the important bondlengths and angles and
put them in a table.
Conclude your report with a short discussion of the general features
of the structure.