- Contact: [email protected]
- WEB SITE
- Ortep-3 - http://www.chem.gla.ac.uk/~louis/software/ortep3/ | [CCP14 UK Mirror | CCP14 Australian Mirror | CCP14 Canadian Mirror]
- CCP14 Based WinGX/Ortep-3 Tutorial Pages
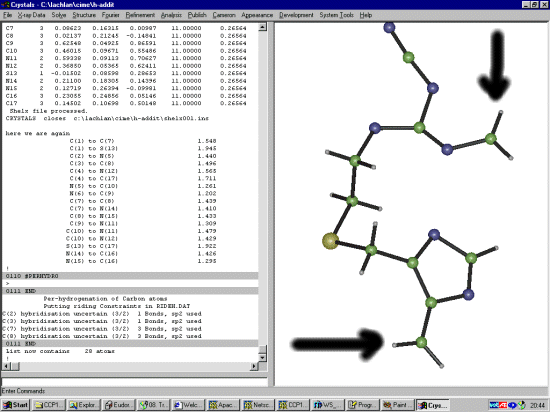
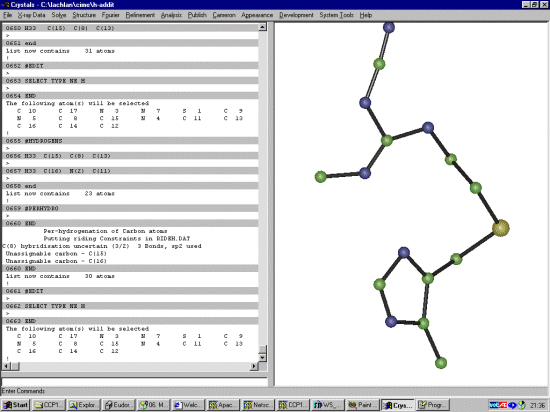
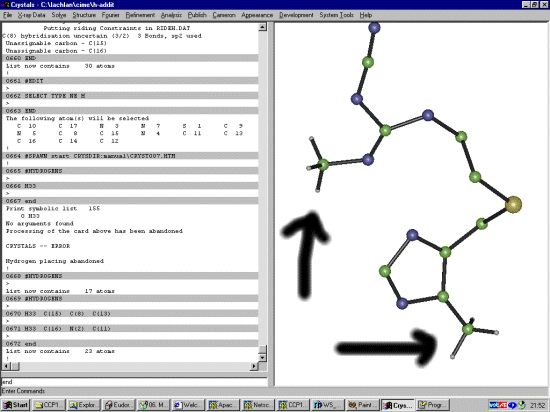
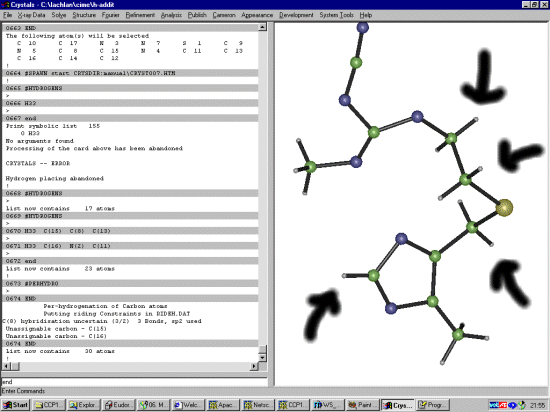
The Shelx file probably does not have the SFAC for Hydrogen so it would be good to add this in now. Manually edit the Shelx file and change:
SFAC S N C UNIT 1 6 10to:
SFAC S N C H UNIT 1 6 10 9
You may like to copy this file into a new directory before running Crystals (or any other single crystal suite)